热图
 1.解释图片特点
1.解释图片特点
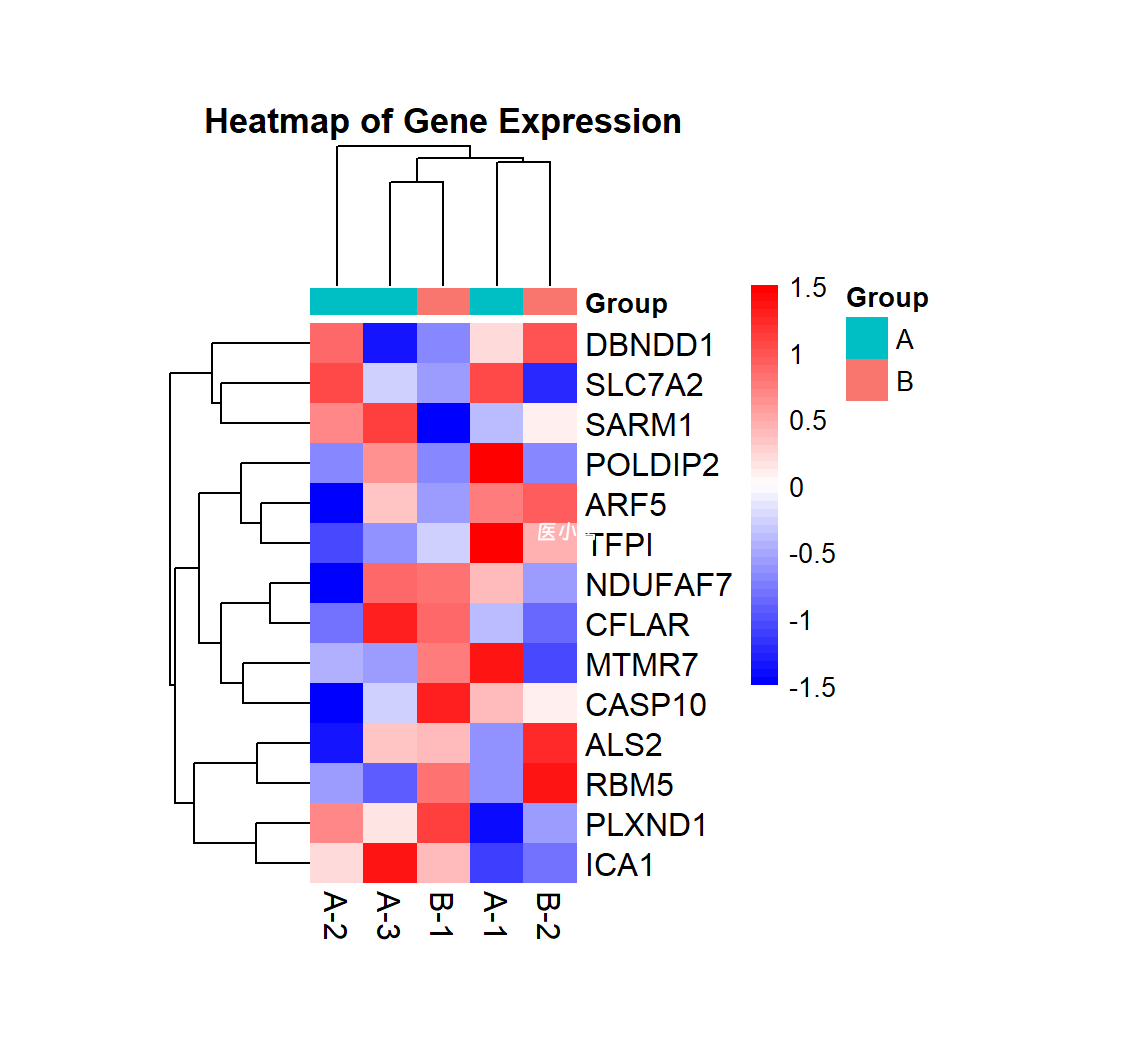
这张图是一个热图(Heatmap),用于展示不同样本之间的基因表达量的差异。该图的特点如下:
数据分组:样本被分为两个组(A 和 B),每组的样本通过不同的颜色条标记,分别为青色和粉色,显示在图的顶部。
基因名称:右侧的标签列显示了每个基因的名称。
颜色梯度:图中使用了从蓝色到红色的颜色梯度表示基因表达量的变化,蓝色代表较低表达(-1.5以下),红色代表较高表达(1.5以上),颜色的强度表示基因表达的水平。
聚类树:在行和列的两侧都有聚类树,表示基因和样本之间的相似性。
图例:右侧包含颜色梯度的图例,表示数据的范围和两个分组的名称。
2.数据要求
为了生成类似的热图,数据需要满足以下要求:
数据矩阵:一个包含多个基因(行)和多个样本(列)的矩阵。每个单元格表示基因在某个样本中的表达量。
样本分组信息:样本被分为两个或多个组,需为每个样本标记其所属的组(如组A、组B)。
数值范围:基因表达量应该是连续数据,范围应覆盖正负值(以突出高表达和低表达之间的差异)。
聚类分析:用于行和列的聚类,展示基因和样本之间的相似性。
3.用R代码生成虚拟数据并绘制热图
# 生成虚拟数据
set.seed(123)
gene_names <- c("SLC7A2", "PLXND1", "MTMR7", "ARF5", "SARM1", "POLDIP2", "NDUFAF7",
"ICA1", "CFLAR", "ALS2", "TFPI", "CASP10", "DBNDD1", "RBM5")
sample_names <- c("A-1", "A-2", "A-3", "B-1", "B-2")
group <- c("A", "A", "A", "B", "B")
data_matrix <- matrix(rnorm(14 * 5, mean = 0, sd = 1), nrow = 14, ncol = 5, dimnames = list(gene_names, sample_names))
data_frame <- as.data.frame(data_matrix)
# 添加分组信息到列注释数据框
group_info <- data.frame(Group = group)
rownames(group_info) <- sample_names
# 保存为CSV文件
write.csv(data_frame, file = "heatmap_data.csv", row.names = TRUE)
write.csv(group_info, file = "group_info.csv", row.names = TRUE)
# 读取CSV文件
heatmap_data <- read.csv("heatmap_data.csv", row.names = 1)
group_info <- read.csv("group_info.csv", row.names = 1)
group_info <- group_info
heatmap_data <- data_frame
# 使用pheatmap绘制热图
if (!require("pheatmap")) install.packages("pheatmap")
library(pheatmap)
library(RColorBrewer)
# 分离基因表达数据和分组信息
expression_data <- heatmap_data[, -ncol(heatmap_data)]
# 设置颜色
ann_colors <- list(Group = c(A = "#66c2a5", B = "#fc8d62"))
# 按列聚类
pheatmap(heatmap_data,
cluster_rows = TRUE,
cluster_cols = TRUE,
annotation_col = group_info,
annotation_colors = list(Group = c(A = "#00BFC4", B = "#F8766D")),
color = colorRampPalette(c("blue", "white", "red"))(50),
scale = "row",
fontsize_row = 12,
fontsize_col = 12,
legend_breaks = seq(-1.5, 1.5, by = 0.5),
legend_labels = seq(-1.5, 1.5, by = 0.5),
annotation_legend = TRUE,
main = "Heatmap of Gene Expression",
border_color = NA,
cellwidth = 20,
cellheight = 15)
# 分组展示
pheatmap(heatmap_data,
cluster_rows = TRUE,
cluster_cols = FALSE,
annotation_col = group_info,
annotation_colors = list(Group = c(A = "#00BFC4", B = "#F8766D")),
color = colorRampPalette(c("blue", "white", "red"))(50),
scale = "row",
fontsize_row = 12,
fontsize_col = 12,
legend_breaks = seq(-1.5, 1.5, by = 0.5),
legend_labels = seq(-1.5, 1.5, by = 0.5),
annotation_legend = TRUE,
main = "Heatmap of Gene Expression",
border_color = NA,
cellwidth = 20,
cellheight = 15,
clustering_distance_cols = "euclidean",
clustering_method = "ward.D2",
gaps_col = c(3))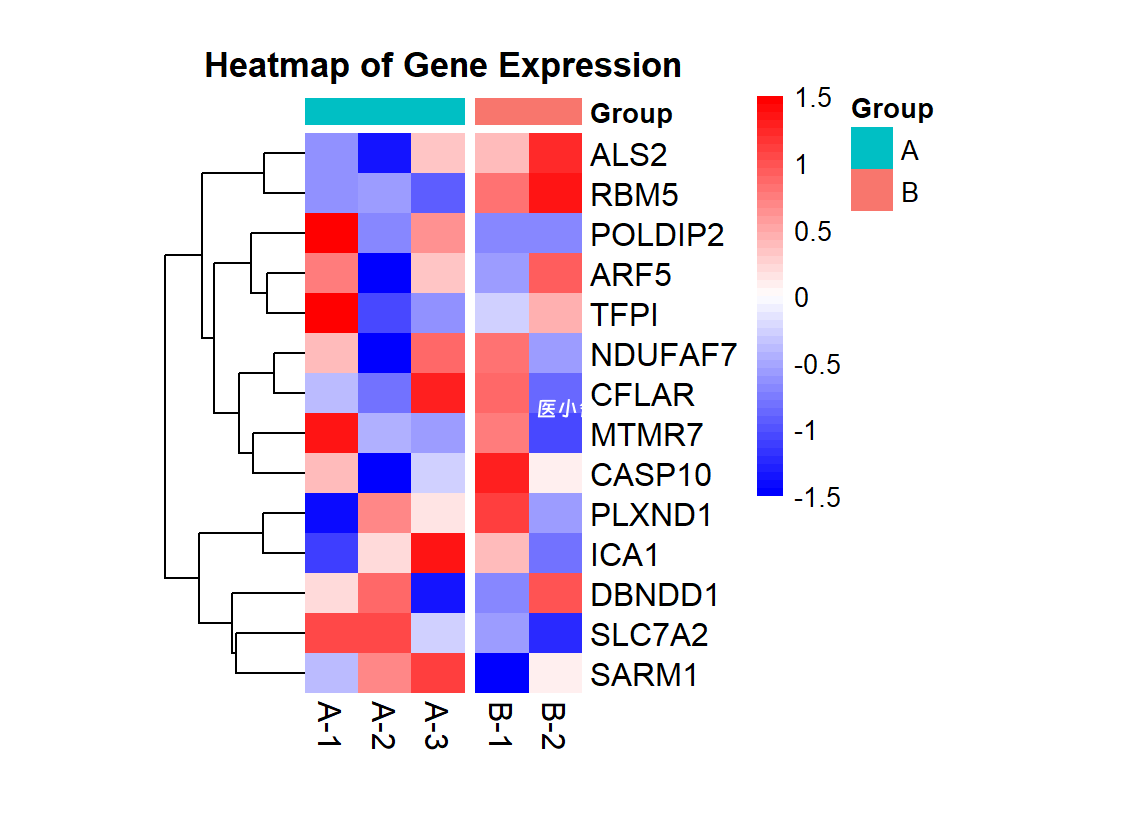
阅读剩余
版权声明:
作者:Q
链接:https://yunshangtulv.com.cn/?p=802
文章版权归作者所有,未经允许请勿转载。
THE END